
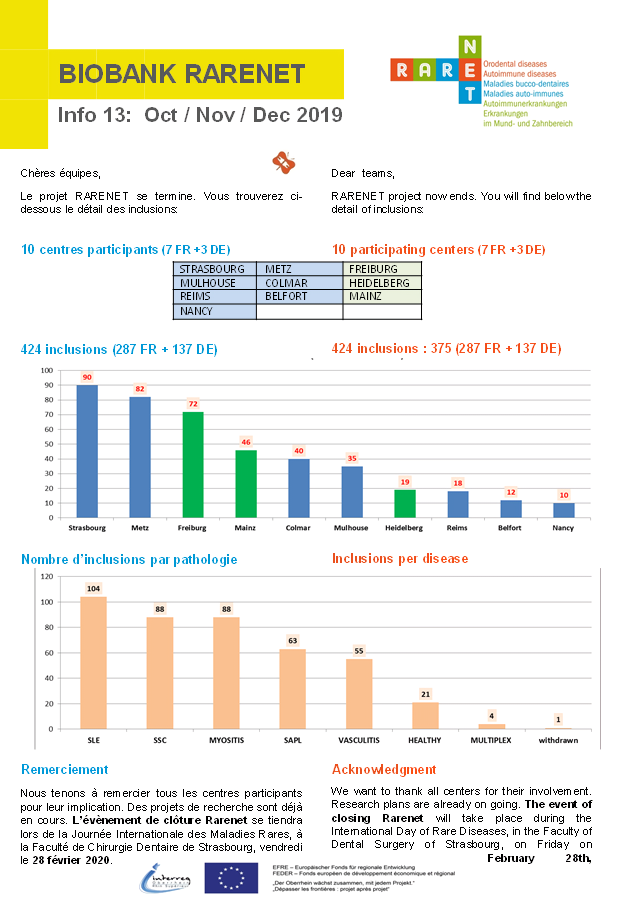
Biobanque RARENET Oct/ Nov/ Dec 2019
The management of lymphoma in patients with primary immunodeficiency (PID) is challenging because of its poor prognosis and complex therapeutic approaches. We conducted a systematic literature review of case-reports, case-series, and cohorts indexed in MEDLINE reporting the association of lymphoma and PID. One hundred and eighty-two articles were selected out of 787. We identified 386 cases. Median age at diagnosis of PID and lymphoma was 9.5 and 12 years old, respectively. T-cell deficiencies were the main PIDs associated with lymphoma (57%). The most prevalent lymphoma was diffuse large B-cell lymphoma (33.5%). Epstein-Barr Virus-driven lymphomas were mostly observed in innate immunodeficiencies (when reported). Complete response to treatment was observed in 65.8% of the cases. Death occurred in 38.2%. Few allogenic stem cell transplantations were performed (29 cases). Our detailed analysis of the literature provides a landscape of lymphoma’s occurrence in PID. Devoted studies in specific sub-groups of patients at risk are needed to develop dedicated protocols.
Read the article: https://www.ncbi.nlm.nih.gov/pubmed/31580160
Type I interferons are highly potent cytokines essential for self-protection against tumors and infections. Deregulations of type I interferon signaling are associated with multiple diseases that require novel therapeutic options. Here, we identified the small molecule, IT1t, a previously described CXCR4 ligand, as a highly potent inhibitor of Toll-like receptor 7 (TLR7)-mediated inflammation. IT1t inhibits chemical (R848) and natural (HIV) TLR7-mediated inflammation in purified human plasmacytoid dendritic cells from blood and human tonsils. In a TLR7-dependent lupus-like model, in vivo treatment of mice with IT1t drives drastic reduction of both systemic inflammation and anti-double-stranded DNA autoantibodies and prevents glomerulonephritis. Furthermore, IT1t controls inflammation, including interferon α secretion, in resting and stimulated cells from patients with systemic lupus erythematosus. Our findings highlight a groundbreaking immunoregulatory property of CXCR4 signaling that opens new therapeutic perspectives in inflammatory settings and autoimmune diseases.
Read the article: https://www.ncbi.nlm.nih.gov/pubmed/31309143
OBJECTIVES: cardiac involvement is the second most frequent systemic sclerosis (SSc) related cause of death. It remains mostly asymptomatic in the early stage and is underdiagnosed with routine screening. Cardiac magnetic resonance imaging (CMR) could improve cardiac assessment of patients and noteworthily, new sequences allow the detection of diffuse myocardial fibrosis (DMF) by native T1 mapping. The aim of this study was to determine the prevalence of cardiac involvement by CMR native T1 mapping and its correlation with echocardiography data and non-cardiac manifestations in SSc patients.
METHODS: patients fulfilling the ACR/EULAR classification criteria for SSc were prospectively included between 2014 and 2016. They underwent CMR at 1.5T, including native T1 and T2 mapping, and Late Gadolinium Enhancement (LGE) as a part of routine follow up. Routine biological tests (mainly BNP and CRP) were centralized in the hospital laboratory.
RESULTS: seventy-two unselected patients were included. Thirty six patients (50%) had elevated T1 (ET1) (mean T1 1097±14 ms). CMR cardiac functional parameters were similar in ET1 and normal T1 (NT1). Echocardiography was normal in 18 (50%) of ET1. ET1 and NT1 groups were similar for cardiovascular risk factors and ischemic heart disease. ET1 was not correlated with any clinical or echocardiographic parameter or antibody profile. Thirty-six percent of patients with ET1 had no cardiac symptoms, normal echocardiography and CMR LVEF, and no LGE.
CONCLUSION: native T1 mapping detects left ventricular ET1 (potential DMF) in 50% of patients with SSc and a third of them had a normal conventional screening including standard CMR. In the future, further studies are needed to confirm the benefit of use of native T1 mapping as a part of routine follow up to detect earlier pejorative cardiac involvement in SSc patients.
Read the article: https://www.sciencedirect.com/science/article/pii/S004901721830711X?via%3Dihub
Ikaros is a transcription factor with key roles in lymphocyte development and homeostasis. It is encoded by the IKAROS family zinc finger protein 1 (IKZF1) gene, which contains highly conserved N-terminal and C-terminal zinc finger domains and is highly expressed in hematopoietic cells. The functions of Ikaros were first illustrated in different sets of IKZF1-deficient mice. In humans, IKZF1 loss-of-function (LOF) somatic variants are associated with leukemogenesis in B cell acute lymphoblastic leukemia (B-ALL) [1]. More recently, different germline heterozygous variants, acting either by haploinsufficiency or by a dominant negative effect in IKZF1, were identified in autosomal dominant forms of common variable immunodeficiencies (CVID) or combined immunodeficiencies (CID) [2, 3, 4]. A series of families showed a progressive loss of B cells and hypogammaglobulinemia, but the absence of symptoms in several mutated patients suggests an incomplete penetrance. Additional…
Read the article: https://link.springer.com/article/10.1007%2Fs10875-019-00643-2
BACKGROUND: Autosomal dominant gain-of-function mutations in human stimulator of interferon genes (STING) lead to a severe autoinflammatory disease called STING-associated vasculopathy with onset in infancy that is associated with enhanced expression of interferon-stimulated gene transcripts.
OBJECTIVE: The goal of this study was to analyze the phenotype of a new mouse model of STING hyperactivation and the role of type I interferons in this system.
METHODS: We generated a knock-in model carrying an amino acid substitution (V154M) in mouse STING, corresponding to a recurrent mutation seen in human patients with STING-associated vasculopathy with onset in infancy. Hematopoietic development and tissue histology were analyzed. Lymphocyte activation and proliferation were assessed in vitro. STING V154M/wild-type (WT) mice were crossed to IFN-α/β receptor (IFNAR) knockout mice to evaluate the type I interferon dependence of the mutant Sting phenotype recorded.
RESULTS: In STING V154M/WT mice we detected variable expression of inflammatory infiltrates in the lungs and kidneys. These mice showed a marked decrease in survival and developed a severe combined immunodeficiency disease (SCID) affecting B, T, and natural killer cells, with an almost complete lack of antibodies and a significant expansion of monocytes and granulocytes. The blockade in B- and T-cell development was present from early immature stages in bone marrow and thymus. In addition, in vitro experiments revealed an intrinsic proliferative defect of mature T cells. Although the V154M/WT mutant demonstrated increased expression of interferon-stimulated genes, the SCID phenotype was not reversed in STING V154M/WT IFNAR knockout mice. However, the antiproliferative defect in T cells was rescued partially by IFNAR deficiency.
CONCLUSIONS: STING gain-of-function mice developed an interferon-independent SCID phenotype with a T-cell, B-cell, and natural killer cell developmental defect and hypogammaglobulinemia that is associated with signs of inflammation in lungs and kidneys. Only the intrinsic proliferative defect of T cells was partially interferon dependent.
Read the article: https://www.ncbi.nlm.nih.gov/pubmed/29800647
BACKGROUND: Down syndrome (DS) is the most common form of viable chromosomal abnormality. DS is associated with recurrent infections, auto-immunity and malignancies in children. Little is known about immunity and infections in DS at adulthood.
METHODS: We studied two separate group of adults (> 18 years old) with DS in a single referral tertiary center (Strasbourg University Hospital). The first group included 37 ambulatory DS patients between November 2014 and May 2017. We analyzed exhaustive serological and immunobiological parameters, at one point, together with the prevalence of infections, autoimmune manifestations and malignancies. The second group included 64 hospitalized patients (138 stays) in the same center, between January 2005 and December 2016.
RESULTS: One hundred and one adult patients with DS were included. Unlike children and despite a global lymphopenia, adults with DS underwent few infections in our ambulatory group. They did not experience any malignancy and, apart from hypothyroidism, they presented only occasional autoimmune manifestations. Hospitalized DS patients were older than ambulatory ones (median age 47 years (18-73) vs. 27 (18-52), p < 0.0001) and admitted mostly for infections (76.8%). Infections were associated with epilepsy and dementia (OR 6.5 (2.2-19), p = 0.001; p = 0.0006 in multivariate analysis) and higher mortality (OR 7.4 (1.4-37), p = 0.01).
CONCLUSION: Despite persistent immunobiological abnormalities at adulthood, young ambulatory adults with DS remain healthy with a low rate of infections. Infections are associated with neurological degeneration and increase the mortality arguing for a dedicated support of older DS patients.
TRIAL REGISTRATION: ClinicalTrials.gov: NCT01663675 (August 13, 2012). Hospital Clinical Research Program (PHRC): number 2012-A00466-37 (Dr Y. Alembik).
Read the article: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6329099/
Dental anomalies occur frequently in a number of genetic disorders and act as major signs in diagnosing these disorders. We present definitions of the most common dental signs and propose a classification usable as a diagnostic tool by dentists, clinical geneticists, and other health care providers. The definitions are part of the series Elements of Morphology and have been established after careful discussions within an international group of experienced dentists and geneticists. The classification system was elaborated in the French collaborative network “TÊTECOU” and the affiliated O-Rares reference/competence centers. The classification includes isolated and syndromic disorders with oral and dental anomalies, to which causative genes and main extraoral signs and symptoms are added. A systematic literature analysis yielded 408 entities of which a causal gene has been identified in 79%. We classified dental disorders in eight groups: dental agenesis, supernumerary teeth, dental size and/or shape, enamel, dentin, dental eruption, periodontal and gingival, and tumor-like anomalies. We aim the classification to act as a shared reference for clinical and epidemiological studies. We welcome critical evaluations of the definitions and classification and will regularly update the classification for newly recognized conditions.
Read the article: https://www.ncbi.nlm.nih.gov/pubmed/31468724
Amelogenesis imperfecta (AI) is a heterogeneous group of rare inherited diseases presenting with enamel defects. More than 30 genes have been reported to be involved in syndromic or non-syndromic AI and new genes are continuously discovered (Smith et al., 2017). Whole-exome sequencing was performed in a consanguineous family. The affected daughter presented with intra-uterine and postnatal growth retardation, skeletal dysplasia, macrocephaly, blue sclerae, and hypoplastic AI. We identified a homozygous missense mutation in exon 11 of SLC10A7 (NM_001300842.2: c.908C>T; p.Pro303Leu) segregating with the disease phenotype. We found that Slc10a7 transcripts were expressed in the epithelium of the developing mouse tooth, bones undergoing ossification, and in vertebrae. Our results revealed that SLC10A7 is overexpressed in patient fibroblasts. Patient cells display altered intracellular calcium localization suggesting that SLC10A7 regulates calcium trafficking. Mutations in this gene were previously reported to cause a similar syndromic phenotype, but with more severe skeletal defects (Ashikov et al., 2018;Dubail et al., 2018). Therefore, phenotypes resulting from a mutation in SLC10A7 can vary in severity. However, AI is the key feature indicative of SLC10A7 mutations in patients with skeletal dysplasia. Identifying this important phenotype will improve clinical diagnosis and patient management.
Read the article: https://www.frontiersin.org/articles/10.3389/fgene.2019.00504/full
Cathepsin C (CatC) is a cysteine protease involved in a variety of immune and inflammatory pathways such as activation of cytotoxicity of various immune cells. Homozygous or compound heterozygous variants in the CatC coding gene CTSC cause different conditions that have in common severe periodontitis. Periodontitis may occur as part of Papillon-Lefèvre syndrome (PLS; OMIM#245000) or Haim-Munk syndrome (HMS; OMIM#245010), or may present as an isolated finding named aggressive periodontitis (AP1; OMIM#170650). AP1 generally affects young children and results in destruction of the periodontal support of the primary dentition. In the present study we report exome sequencing of a three generation consanguineous Turkish family with a recessive form of early-onset AP1. We identified a novel homozygous missense variant in exon 2 of CTSC (NM_148170, c.G302C, p.Trp101Ser) predicted to disrupt protein structure and to be disease causing. This is the first described CTSC variant specific to the nonsyndromic AP1 form. Given the broad phenotypic spectrum associated with CTSC variants, reporting this novel variant gives new insights on genotype/phenotype correlations and might improve diagnosis of patients with early-onset AP1.
Read the article: https://www.ncbi.nlm.nih.gov/pubmed/31068678